Histone Modifications Alter Chromatin Structure
The structure of chromatin fluctuates in concert with the stages of the cell cycle: during interphase the chromatin is structurally loose to allow access to RNA and DNA polymerases that transcribe and replicate the DNA. The local structure of chromatin during interphase depends on the genes present on the DNA: DNA coding genes that are actively transcribed ("turned on") are more loosely packaged and are found associated with RNA polymerases (referred to as euchromatin) while DNA coding inactive genes ("turned off") are found associated with structural proteins and are more tightly packaged (heterochromatin). As the cell prepares to divide, i.e. enters mitosis or meiosis, the chromatin packages more tightly to facilitate segregation of the chromosomes during anaphase. During this stage of the cell cycle this makes the individual chromosomes in many cells visible by optical microscope.
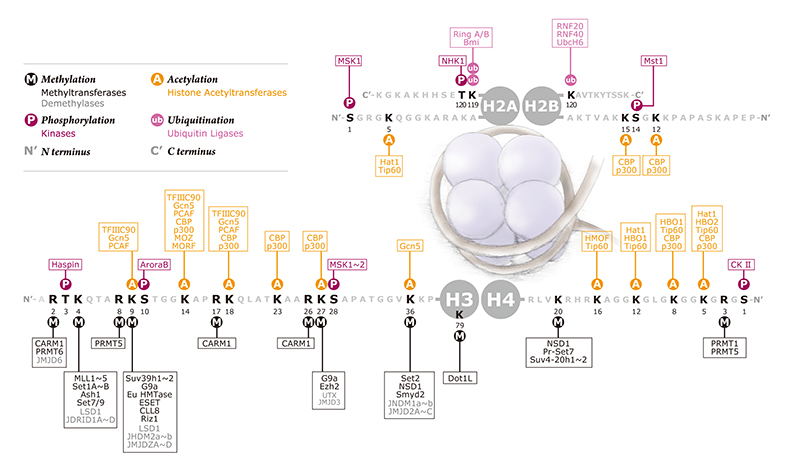
Chromatin is comprised of nucleosome repeats with themselves are comprised of 147bp of dsDNA wrapped 1.67 left-handed superhelical turns around a histone octomer of two H3-H4 dimers bound to two H2A-H2B dimers. Changes in local or global chromatin structure occur through epigenetic chemical modifications imposed upon histone proteins to alter DNA packing. Histones contain positively charged residues (predominantly lysines) in their protruding tails which interact with the negative charged of the dsDNA backbone to facilitate an electrostatic interaction that maintains wrapped DNA around the nucleosome. Covalent modifications imposed on histone tails alter the strength of the interaction with the dsDNA backbone, leading to altered states of gene expression and genetic events, including chromatin condensation, repair, and recombination. Such modifications include ubiquitylation, sumoylation, phosphorylation, acetylation, carbonylation, amd methylation.
Overall, these modifications achieve two goals:
1) alteration of higher order chromatin structure via alterations of nucleosome interactions
2) Recruitment of non-histone proteins to chromatin (see below)
Histone acetylation and phosphorylation reduce the positive charge of histones to disrupt the electrostatic interactions between histones (+ charge) and DNA (- charge). Histone acetylation neutralizes the positive charge of the histone tail to diminish affinity for the proximal negatively charged DNA strand. This leads to a less compact "loose" chromatin structure that facilitates DNA accessibility by transcriptional machinery. Acetylation occurs on a number of histone tail lysines (positively charged residue) including H3K9, H3K14, H3K18, H4K5, H4K8, and H4K12. Histone acetylations are commonly enriched at enhancer elements and gene promoters to facilitate access for transcription factors.
Histone acetyl transferases (HATs) are the family of enzymes responsible for transferring acetyl groups from acetyl-coenzyme A (acetyl CoA) to positively-charged target residues (i.e. lysines) on histone tails such as H3K9, H3K18, and H3K27. Examples include CBP-p300 that acetylate H3K18 and H3K27. Histone de-acetylases (HDACs) catalyze the removal of the acetyl groups from histone tails to re-compact the chromatin. HDACs are grouped into four classes, and are recruited to DNA via co-repressor complexes that remodel chromatin in an ATP-dependent manner. For example, the two major class I HDAC co-repressor complexes are Sin3 and NuRD which contain HDAC1 and HDAC2.
Histone phosphorylation is site-specific and relatively less common than acetylation. Since phosphates add negative charge to histone tails, this would be expected to lead to less compact chromatin structure as the charge should neutralize or even repel from the negatively charged DNA backbone. However, the genome-wide phosphorylation of H3S10 that marks mitosis is associated with chromatin becoming more condensed. This counter-intuitive observation may be explained by displacement of heterochromatin protein 1 (HP1) during metaphase that allows for chromatin remodelling and attachment to mitotic spindle.
Histone methylation occurs primarily on the side-chains of lysines and arginines of histone H3 and histone H4. Methylation does not alter the charge of the histone, and can occur in successive magnitude as mono-, di-, or tri- methylation. More specifically, lysine residues can be mono-, di-, or tri- methylated, while arginine residues can be mono- or di-methylated. When certain residues are methylated they hold DNA together strongly and restrict access to various enzymes. Since methylation can either compact or loosen chromatin, an empirically derived histone code has emerged to classify the effects of these differential methylations. For example, the best characterized methylation marks associated with epigenetic chromatin silencing include H3K9Me3, and H4K20Me3. In contrast, other histone methylation marks are associated with open chromatin, such as H3K4me3.
Histone methyl transferases (HMTs) methylate target lysines or arginines. For example, in mammals, the HMT SUV39H1 imposes the H3K9Me3 mark associates with HP1 recruitment and gene silencing, and constitutive heterochromatin formation. SUV4-20H1 and SUV4-20H2 impose the H4K20Me3 mark also associated with trancriptional repression and heterochromatin formation.
Other histone modifications that alter epigenetic status include ubiquitylation, sumoylation, and deimination.
Chromatin is comprised of nucleosome repeats with themselves are comprised of 147bp of dsDNA wrapped 1.67 left-handed superhelical turns around a histone octomer of two H3-H4 dimers bound to two H2A-H2B dimers. Changes in local or global chromatin structure occur through epigenetic chemical modifications imposed upon histone proteins to alter DNA packing. Histones contain positively charged residues (predominantly lysines) in their protruding tails which interact with the negative charged of the dsDNA backbone to facilitate an electrostatic interaction that maintains wrapped DNA around the nucleosome. Covalent modifications imposed on histone tails alter the strength of the interaction with the dsDNA backbone, leading to altered states of gene expression and genetic events, including chromatin condensation, repair, and recombination. Such modifications include ubiquitylation, sumoylation, phosphorylation, acetylation, carbonylation, amd methylation.
Overall, these modifications achieve two goals:
1) alteration of higher order chromatin structure via alterations of nucleosome interactions
2) Recruitment of non-histone proteins to chromatin (see below)
Histone acetylation and phosphorylation reduce the positive charge of histones to disrupt the electrostatic interactions between histones (+ charge) and DNA (- charge). Histone acetylation neutralizes the positive charge of the histone tail to diminish affinity for the proximal negatively charged DNA strand. This leads to a less compact "loose" chromatin structure that facilitates DNA accessibility by transcriptional machinery. Acetylation occurs on a number of histone tail lysines (positively charged residue) including H3K9, H3K14, H3K18, H4K5, H4K8, and H4K12. Histone acetylations are commonly enriched at enhancer elements and gene promoters to facilitate access for transcription factors.
Histone acetyl transferases (HATs) are the family of enzymes responsible for transferring acetyl groups from acetyl-coenzyme A (acetyl CoA) to positively-charged target residues (i.e. lysines) on histone tails such as H3K9, H3K18, and H3K27. Examples include CBP-p300 that acetylate H3K18 and H3K27. Histone de-acetylases (HDACs) catalyze the removal of the acetyl groups from histone tails to re-compact the chromatin. HDACs are grouped into four classes, and are recruited to DNA via co-repressor complexes that remodel chromatin in an ATP-dependent manner. For example, the two major class I HDAC co-repressor complexes are Sin3 and NuRD which contain HDAC1 and HDAC2.
Histone phosphorylation is site-specific and relatively less common than acetylation. Since phosphates add negative charge to histone tails, this would be expected to lead to less compact chromatin structure as the charge should neutralize or even repel from the negatively charged DNA backbone. However, the genome-wide phosphorylation of H3S10 that marks mitosis is associated with chromatin becoming more condensed. This counter-intuitive observation may be explained by displacement of heterochromatin protein 1 (HP1) during metaphase that allows for chromatin remodelling and attachment to mitotic spindle.
Histone methylation occurs primarily on the side-chains of lysines and arginines of histone H3 and histone H4. Methylation does not alter the charge of the histone, and can occur in successive magnitude as mono-, di-, or tri- methylation. More specifically, lysine residues can be mono-, di-, or tri- methylated, while arginine residues can be mono- or di-methylated. When certain residues are methylated they hold DNA together strongly and restrict access to various enzymes. Since methylation can either compact or loosen chromatin, an empirically derived histone code has emerged to classify the effects of these differential methylations. For example, the best characterized methylation marks associated with epigenetic chromatin silencing include H3K9Me3, and H4K20Me3. In contrast, other histone methylation marks are associated with open chromatin, such as H3K4me3.
Histone methyl transferases (HMTs) methylate target lysines or arginines. For example, in mammals, the HMT SUV39H1 imposes the H3K9Me3 mark associates with HP1 recruitment and gene silencing, and constitutive heterochromatin formation. SUV4-20H1 and SUV4-20H2 impose the H4K20Me3 mark also associated with trancriptional repression and heterochromatin formation.
Other histone modifications that alter epigenetic status include ubiquitylation, sumoylation, and deimination.
Histone modifications recruit non-histone proteins to chromatin
Histone modifications recruit non-histone proteins to chromatin by establishing compatible interaction surfaces on chromatin which are specific for certain domains found within the target protein. Often these proteins further remodel chromatin to execute step-wise changes in gene expression in accordance with a given signaling cascade. Conversely some modifications prevent docking of non-histone proteins onto chromatin. Below is a summary of corresponding protein domains which recognize specific modifications.
Methylation - recognized by Chromo-like domains of the 'Royal' family: chromo, tudor, MBT and non-related PHD domains
- recognized by chromo domain (Ex. CHD1 - ATPase, HP1 - deacetylase/methyltransferase, PC2 - polycomb protein 2 - ubiquitin ligase for H2A)
- recognized by Tudor domain (ex. JMJD2A - histone lysine demethylase)
- recognized by PHD domains (BPTF from NURF complex, ING2 from mSin3a-HDAC1 complex)
Acetylation - recognized by bromo domains
Phosphorylation - recognized by domain within 14-3-3 proteins
Methylation - recognized by Chromo-like domains of the 'Royal' family: chromo, tudor, MBT and non-related PHD domains
- recognized by chromo domain (Ex. CHD1 - ATPase, HP1 - deacetylase/methyltransferase, PC2 - polycomb protein 2 - ubiquitin ligase for H2A)
- recognized by Tudor domain (ex. JMJD2A - histone lysine demethylase)
- recognized by PHD domains (BPTF from NURF complex, ING2 from mSin3a-HDAC1 complex)
Acetylation - recognized by bromo domains
Phosphorylation - recognized by domain within 14-3-3 proteins
