X-ray Crystallography: A working man's intro
One of the most pressing issues in cancer therapy is development of chemoresistance. This may be achieved through several mechanisms. For instance, cancer cells select for DNA polymerases that can replicate over cisplatin, a platinum-based chemotherapeutic which induces lesions in DNA that induce interstrand crosslinks. Selection for low fidelity polymerases allows cancer cells to evade DNA repair or removal checks. Structural biologists want to understand how such enzymes can recognise the cisplatin lesion. What is the active site chemistry of the interaction? Such knowledge could direct design of other chemotherapeutics that this polymerase cannot replicate over.
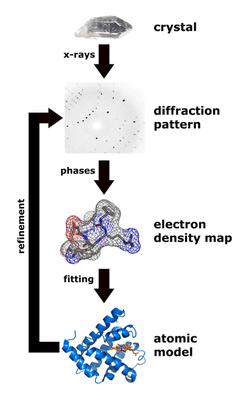
A crystal structure of such a low fidelity polymerase would provide information required to design a drug that can interact with it. To get the crystal structure, you need to purify the protein. In fact, around 90% of a structural biologist's time is spent trying to purify proteins to 'grow' crystals for analysis. To derive a three-dimensional picture of all the atoms in a molecule like an enzyme, the most powerful technique to use is X-ray crystallography. X-ray crystallography uses the interaction of X-ray photons with electrons in a molecule. X-rays (photons) are literally bombarded against the crystal and are diffracted off the electron 'clouds' surrounding atoms. Collecting these diffraction patterns is a task in itself. In order to resolve the distances between atoms, you need a means to measure the waves between atoms, which are separated by distances of around 1 angstrom, a hundred-billionth of a meter. There is currently no such thing as an X-ray microscope because of the difficulty in making a lens that can capture the tiny X-ray waves.
This is why a "crystal" is needed. Diffraction patterns from millions of molecules are used because patterns can't be reconstructed through a lens. Researchers need to grow crystals, because the atoms are all orientated in the same way. Millions of molecules are needed to get enough 'signal' for the structural information. Owing to the lens problem, half of the information (phases of light) is lost, so researchers need to run computer programmes to get the lost phases back. Another difficult job. In fact, this is often a PhD thesis!
Once diffracted waves are run through a computer programme, researchers get the electron density distribution and model the protein structure, something which is now done at synchrotrons. A synchrotron is an enormous electron accelerator, which is used to produce high-intensity X-ray beams. There is one in Grenoble, an impressive 1 km across, which cost over a billion euros to build. Synchrotrons "accelerate electrons to almost the speed of light."
A crystal structure of such a low fidelity polymerase would provide information required to design a drug that can interact with it. To get the crystal structure, you need to purify the protein. In fact, around 90% of a structural biologist's time is spent trying to purify proteins to 'grow' crystals for analysis. To derive a three-dimensional picture of all the atoms in a molecule like an enzyme, the most powerful technique to use is X-ray crystallography. X-ray crystallography uses the interaction of X-ray photons with electrons in a molecule. X-rays (photons) are literally bombarded against the crystal and are diffracted off the electron 'clouds' surrounding atoms. Collecting these diffraction patterns is a task in itself. In order to resolve the distances between atoms, you need a means to measure the waves between atoms, which are separated by distances of around 1 angstrom, a hundred-billionth of a meter. There is currently no such thing as an X-ray microscope because of the difficulty in making a lens that can capture the tiny X-ray waves.
This is why a "crystal" is needed. Diffraction patterns from millions of molecules are used because patterns can't be reconstructed through a lens. Researchers need to grow crystals, because the atoms are all orientated in the same way. Millions of molecules are needed to get enough 'signal' for the structural information. Owing to the lens problem, half of the information (phases of light) is lost, so researchers need to run computer programmes to get the lost phases back. Another difficult job. In fact, this is often a PhD thesis!
Once diffracted waves are run through a computer programme, researchers get the electron density distribution and model the protein structure, something which is now done at synchrotrons. A synchrotron is an enormous electron accelerator, which is used to produce high-intensity X-ray beams. There is one in Grenoble, an impressive 1 km across, which cost over a billion euros to build. Synchrotrons "accelerate electrons to almost the speed of light."

Although there are several synchrotrons (electron accelerators) in Europe, you need to apply for time to use them. This is often funded by a grant for one day's worth of measurement. Researchers make regular trips to Grenoble carrying little 0.1mm wide crystals in liquid nitrogen. The one disadvantage of the X-ray method of deciphering protein structure is that researchers need lots of protein, on average around 10mg of purified protein! Also, you often want to verify X-ray crystallographic models by complementary analysis. For instance, electron-microscopy is useful, because you need 100-fold less protein.
Through deciphering the molecular architecture of the cell, researchers can envision the chemistry of DNA repair, and utilize the structural models to design enzymatic inhibitors.
Through deciphering the molecular architecture of the cell, researchers can envision the chemistry of DNA repair, and utilize the structural models to design enzymatic inhibitors.
